New Psychedelic Drugs With AI/Machine Learning, Using Searchable Clandestine Chemistry Data


Until the 2010s, chemists like Papa Shulgin and others had to take random guesses at new molecules in a highly-labour-intensive process which could take years. It was paper, and pen. And imagination. In the age of networking cloud computing, machine-learning (ML) can create thousands of suggestions in a matter of milliseconds, and rank them by probability/confidence. For Googling purposes, this whole area is known as cheminformatics.
Big Pharma has been on the case since IBM's Watson became a better diagnostic tool than House, MD. It's only a matter of time until some bright spark figures out we can do it with phenethylamines, tryptamines, opiates, cannabinoids, and the other psychoactive classes.
You can't use AWS, but you certainly can rig up a few Raspberry Pi units into a home cloud network. If you're a developer curious about how to store molecules in a database, this is going to be a fun article to learn from.
Finding Chemistry Data To Experiment With
First, above all things, we're going to need data.
Generic/Library
Now if we just want to play with chemistry data, eMolecules (https://search.emolecules.com/info/products-data-downloads.html) offers a dataset of over 1.1M compounds we can use for reference information. So does IUPAC: https://goldbook.iupac.org/ .
Browse the file tree here:
https://downloads.emolecules.com/free/
And of course, the enormous DrugBank:
https://go.drugbank.com/
This will become important later, for our ML training.
Speaking of training, we're going to need t bookmark this:
https://github.com/LeeJunHyun/The-Databases-for-Drug-Discovery
Querying PUG (PubChem API)
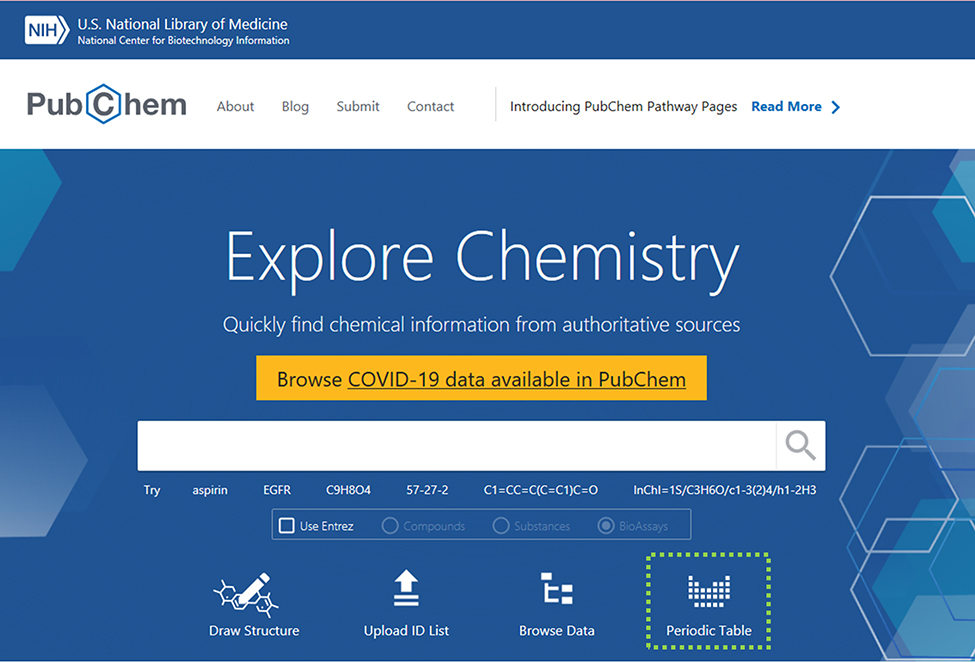
For individual queries, PubChem is our industry standard, which also has a REST API called PUG. All of its records are stored with a CID, or Chemical ID. The rate limit is 5 requests/sec.
- https://pubchem.ncbi.nlm.nih.gov/
- https://pubchem.ncbi.nlm.nih.gov/source/
- https://pubchemdocs.ncbi.nlm.nih.gov/pug-rest-tutorial
To find a list of synonyms:
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/name/mdma/synonyms/XML
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/name/mdma/synonyms/JSON
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/name/3,4-Methylenedioxymethamphetamine/synonyms/XML
To get all data by name:
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/name/mdma/XML
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/name/mdma/JSON
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/name/3,4-Methylenedioxymethamphetamine/JSON
Why not search by formula?
Back to school, you.
Because molecular formulae are not unique. Go to ChemSpider (e.g. https://www.chemspider.com/Search.aspx?rid=fca6125e-747c-4f76-b103-8ea1ce7273b5), and type in C11H15NO2 as your search.
Found 14760 results
Psychoactives
But for the real goods, we want data sources for psychoactive compounds. For most of these, we're going to need to compile them ourselves as a spreadsheet we can export as a CSV file.
Erowid has a massive reference library: https://www.erowid.org/library/books/a_books.shtml
https://www.erowid.org/chemicals/
All in all, we're looking at < 15,000 records, with wildly different effects.
For general governmental data (UN, EU, DEA etc):
- https://www.un-ilibrary.org/content/periodicals/24118338
- https://www.emcdda.europa.eu/system/files/publications/13464/20205648_TD0320796ENN_PDF_rev.pdf
- https://www.google.com/books/edition/Novel_Psychoactive_Substances/qsOcp5LTH-UC?hl=en
- https://www.emcdda.europa.eu/publications_en
- https://www.jneurosci.org/content/34/46/15150
First up, Papa Shulgin's master works, PIHKAL and TIKHAL:
- https://erowid.org/library/books_online/pihkal/pihkal.shtml
- https://erowid.org/library/books_online/tihkal/tihkal.shtml
For other psychedelics, we can parse out David Nichols' work at Purdue from the 1990s:
- https://pubs.acs.org/doi/pdf/10.1021/jm00105a043
- https://pubs.acs.org/doi/abs/10.1021/jm970219x
- https://publications.rwth-aachen.de/record/795276
- https://pubs.acs.org/doi/abs/10.1021/jm00248a004
- https://en.wikipedia.org/wiki/Substituted_benzofuran
Or use structured lists of substituted psychedelic amphetamines:
- https://en.wikipedia.org/wiki/Substituted_amphetamine
- https://en.wikipedia.org/wiki/Substituted_methylenedioxyphenethylamine
- https://en.wikipedia.org/wiki/Substituted_phenethylamine
- https://en.wikipedia.org/wiki/Substituted_phenylmorpholine
For cocaine (phenyltropane) analogues, we can use Robert Clarke and Satendra Singh's work, if you've got the time to list them all out:
- https://pubs.acs.org/doi/abs/10.1021/jm00269a600
- https://www.erowid.org/archive/rhodium/pdf/cocaineanalogs.pdf#45
- https://en.wikipedia.org/wiki/List_of_cocaine_analogues
For cathinones and aminorex, we have hundreds:
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4043811/
- https://en.wikipedia.org/wiki/Substituted_cathinone
- https://en.wikipedia.org/wiki/List_of_aminorex_analogues
For cannabinoids, we can use John Huffman, Alexandros Makriyannis, Pfizer, and Raphael Mechoulam's work:
- https://en.wikipedia.org/wiki/List_of_JWH_cannabinoids
- https://en.wikipedia.org/wiki/List_of_CP_cannabinoids
- https://en.wikipedia.org/wiki/List_of_AM_cannabinoids
- https://en.wikipedia.org/wiki/List_of_HU_cannabinoids
- https://en.wikipedia.org/wiki/Synthetic_cannabinoids
- https://en.wikipedia.org/wiki/List_of_miscellaneous_designer_cannabinoids
The amazing Isomer Design (https://isomerdesign.com/PiHKAL/?domain=tk) lists 13,394 substances of interest. It's a little out of data, but we can roughly put the ongoing list at 15-20,000, with 300-900 being added each year.
No, we're not generating new fentanyls or other dirty compounds.
Identifying & Representing Chemicals As Text
Let's break down how we represent a molecule, using MDMA as an example.
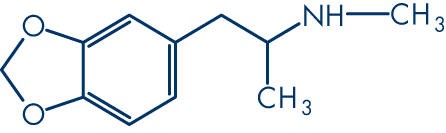
First, we have its common name: MDMA. Then we have its formula: C11H15NO2, and its category: phenylethylamines. It might have dozens of synonyms or brand names, for example: Ecstasy or Midomafetamine. None of these things tell us about the chemistry.
It will have a common name for general usage, such as 3,4-Methylenedioxymethamphetamine.
It will have a unique name given by the International Union of Pure and Applied Chemistry (IUPAC): 1-(1,3-benzodioxol-5-yl)-N-methylpropan-2-amine.
It will also have a unique American Chemical Society CAS Registry number, such as 42542-10-9.
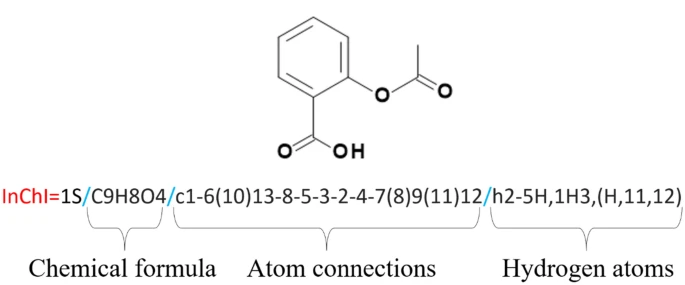
Next, it will have a unique International Chemical Identifier or InChI, such as InChI=1S/C11H15NO2/c1-8(12-2)5-9-3-4-10-11(6-9)14-7-13-10/h3-4,6,8,12H,5,7H2,1-2H3. This will be shortened to a unique hash (key), which appears as so: SHXWCVYOXRDMCX-UHFFFAOYSA-N.
These are all literary and text-searchable strings. But what about the actual physical 2D/3D structure of the molecule?
For that, we use the Simplified Molecular-Input-Line-Entry System (SMILES). From FarLeftiPedia:
The simplified molecular-input line-entry system (SMILES) is a specification in the form of a line notation for describing the structure of chemical species using short ASCII strings. SMILES strings can be imported by most molecule editors for conversion back into two-dimensional drawings or three-dimensional models of the molecules. The original SMILES specification was initiated in the 1980s. It has since been modified and extended. In 2007, an open standard called OpenSMILES was developed in the open-source chemistry community.
The canonical SMILES string for MDMA is CC(CC1=CC2=C(C=C1)OCO2)NC. If you are familiar with geospatial functions in MySQL, PostGIS, ArcGIS etc, it can be thought of conceptually similar to what Well-Known Text (WKT) does for geometry: https://en.wikipedia.org/wiki/Well-known_text_representation_of_geometry .
To summarise, we can store the following data:
- Common name
- Synonyms
- Formula
- International Union of Pure and Applied Chemistry (IUPAC) identifier
- American Chemical Society CAS Registry identifier
- International Chemical Identifier (InChI)
- International Chemical Identifier key (InChIKey)
- SMILES structure
We can directly query SMILES data like SQL, and we can convert it backwards and forwards into molecular vector imagery (e.g. SVG). They can be stored in .MDL (Molfile) format. Many programs exist to deal with this imagery, and they are called molecule editors: https://en.wikipedia.org/wiki/Molecule_editor#Online_editors .
https://pubchem.ncbi.nlm.nih.gov//edit3/index.html --> paste in "CC(CC1=CC2=C(C=C1)OCO2)NC" (without quotes) and hit the return key.
Storing Chemical Molecules In A Database
Any database can store text and index it. However, what we need is specialist functionality for chemistry. One of the first is here: https://homepage.univie.ac.at/norbert.haider/cheminf/moldb6.html

Within the sciences, for some inexplicable reason, database extensions or plugins are referred to as cartridges. A "cartridge" is a cheminformatics toolkit, or a bunch of field types and functions. A database plugin, basically.
tl;dr version: use Postgres & RDKit.
MySQL
You're going to need Mychem: https://mychem.github.io/
Oracle
You're going to need JChem (https://docs.chemaxon.com/display/docs/jchem-cartridge.md) or OrChem (https://orchem.sourceforge.net/)
SQL Server
You're going to need Bingo: https://lifescience.opensource.epam.com/bingo/index.html
PostreSQL
Postgres is the king here. Everything in science seems to use it. Save yourself the time and start with this.
The most popular is RDKit: https://www.rdkit.org/docs/Cartridge.html
But you can also use:
- JChem (https://docs.chemaxon.com/display/docs/jchem-cartridge.md)
- Bingo (https://lifescience.opensource.epam.com/bingo/index.html)
- Sechem (http://bioinfo.uochb.cas.cz/sachem/)
MongoDB
See these:
- https://github.com/mcs07/mongodb-chemistry
- https://matt-swain.com/blog/2014-06-03-chemical-similarity-search-in-mongodb
CouchDB/Elasticsearch
See these:
- https://cjcp.ustc.edu.cn/hxwlxb/en/article/doi/10.1063/1674-0068/31/cjcp1711202
- https://github.com/MysterionRise/pubchem-finder
Docker & Database Setup
Create the database server in the same way you'd use any other and get a copy of DBeaver (https://dbeaver.io/).
Use this image: https://github.com/mcs07/docker-postgres-rdkit
docker run --name mypostgres -p 5432:5432 -e POSTGRES_PASSWORD=mypassword -d mcs07/postgres-rdkitOnce connected in the UI, set up your database:
CREATE DATABASE molecules;
USE molecules;
CREATE EXTENSION IF NOT EXISTS rdkit;
CREATE EXTENSION IF NOT EXISTS "uuid-ossp"Next, set up a table to import your CSV files into, with the new field types and text indices you need.
CREATE TABLE psychoactives (
psy_id bigserial NOT NULL,
psy_uuid uuid NOT NULL DEFAULT uuid_generate_v4(),
src varchar(255) NULL,
family varchar(255) NULL,
label varchar(255) NULL,
common text NULL,
formula varchar(255) NULL,
iupac text NULL,
cas text NULL,
inchi varchar(255) NULL,
inchikey varchar(255) NULL,
structure mol NULL,
fingerprint bfp NULL,
shulgin int2 NULL,
CONSTRAINT psychoactives_pkey PRIMARY KEY (psy_id)
);Note the two rdkit column types here: mol and bfp. The Shulgin column is adapted from the Shulgin Scale (https://en.wikipedia.org/wiki/Shulgin_Rating_Scale).
Next, add a tonne of indices to speed up the text-based querying. IUPAC, CAS, INCHI, INCHIKEY, STRUCTURE, FINGERPRINT should be unique and throw an error if duplicated. This is optional, of course.
CREATE INDEX psychoactives_family_index ON psychoactives USING btree (family);
CREATE INDEX psychoactives_formula_index ON psychoactives USING btree (formula);
CREATE INDEX psychoactives_inchi_index ON psychoactives USING btree (inchi);
CREATE INDEX psychoactives_inchikey_index ON psychoactives USING btree (inchikey);
CREATE INDEX psychoactives_label_index ON psychoactives USING btree (label);
CREATE INDEX psychoactives_psy_uuid_index ON psychoactives USING btree (psy_uuid);
CREATE INDEX psychoactives_shulgin_index ON psychoactives USING btree (shulgin);
CREATE INDEX psychoactives_src_index ON psychoactives USING btree (src);
CREATE INDEX psychoactives_cas_fulltext ON psychoactives USING gin (to_tsvector('english'::regconfig, cas));
CREATE INDEX psychoactives_iupac_fulltext ON psychoactives USING gin (to_tsvector('english'::regconfig, iupac));
CREATE INDEX psychoactives_cas_fulltext ON psychoactives USING gin (to_tsvector('english'::regconfig, common));
CREATE INDEX psychoactives_structure_index ON psychoactives USING gist (structure);We can generate most of the field date from the SMILES data, so all we really need for our CSV data is:
- src
- family
- label
- IUPAC name
- CAS registry #
- structure (SMILES)
An example insert for MDMA:
INSERT INTO psychoactives
(src, family, label, common, iupac, cas, structure)
VALUES
('pikhal','entactogens','MDMA', '3,4-Methylenedioxymethamphetamine', '1-(1,3-benzodioxol-5-yl)-N-methylpropan-2-amine','42542-10-9','CNC(C)Cc1ccc2c(c1)OCO2');And an auto-update from the SMILES data:
UPDATE psychoactives SET
formula = mol_formula (structure),
inchi = mol_inchi (structure),
inchikey = mol_inchikey (structure),
fingerprint = morganbv_fp (structure)Which will create a record like so:
|psy_id|psy_uuid|src|family|label|common|formula|iupac|cas|inchi|inchikey|structure|fingerprint|shulgin|
|------|--------|---|------|-----|-------|-----|---|-----|--------|---------|-----------|-------|
|1|56c0b4d9-8dbb-4731-8629-db5d6c21c142|pikhal|entactogens|MDMA|3,4-Methylenedioxymethamphetamine|C11H15NO2|1-(1,3-benzodioxol-5-yl)-N-methylpropan-2-amine|42542-10-9|InChI=1S/C11H15NO2/c1-8(12-2)5-9-3-4-10-11(6-9)14-7-13-10/h3-4,6,8,12H,5,7H2,1-2H3|SHXWCVYOXRDMCX-UHFFFAOYSA-N|CNC(C)Cc1ccc2c(c1)OCO2|\x02080000020000000080010000800000010001000000020001004004040000000001000800004000a000020010080a0000004040000000024000000004840000|3|
For a much deeper understanding, papers like these from the Journal of Cheminformatics are essential reading:
We present here some of the most popular electronic molecular and macromolecular representations used in drug discovery, many of which are based on graph representations. Furthermore, we describe applications of these representations in AI-driven drug discovery. Our aim is to provide a brief guide on structural representations that are essential to the practice of AI in drug discovery. This review serves as a guide for researchers who have little experience with the handling of chemical representations and plan to work on applications at the interface of these fields.
"Molecular representations in AI-driven drug discovery: a review and practical guide"
https://jcheminf.biomedcentral.com/articles/10.1186/s13321-020-00460-5#additional-information
Querying SMILES Data For Compound Features & Chemical Similarity
It's simple to query text in all the different databases, and particularly useful in Elasticsearch. In Postgres you would do it using standard ts functions.
These create lexemes or tokens to search for.
SELECT to_tsvector('english', phraseto_tsquery('english', 'benzodioxol')::text)
# Result
# 'benzodioxol':1For TEXT columns with a FULLTEXT index, we could search the iupac column for benzodioxol easily:
SELECT *,
ts_rank ('benzodioxol', to_tsquery('english', phraseto_tsquery('english', 'benzodioxol')::text)) AS score,
ts_headline ('benzodioxol', to_tsquery('english', phraseto_tsquery('english', 'benzodioxol')::text)) AS highlighted
FROM psychoactives
WHERE iupac @@ to_tsquery('english', phraseto_tsquery('english', 'benzodioxol')::text)
ORDER BY score DESCWhere this gets interesting is using the SMILES field. All of the rdkit functions are documented here, and they're ugly.
The best way to understand SMILES, as pointed out earlier, if you're not a chemist, is to see it a bit like PostGIS. The shapes are stored as text, accelerated with an index, and queried spatially as a graph.
In all of these examples, our SMILES field (type: mol) is named structure.
Is it valid syntax?
SELECT is_valid_smiles (mol_to_smiles(structure)) FROM psychoactives;Convert to SMARTS
SELECT mol_to_smarts (structure) FROM psychoactives;Convert to SVG
SELECT mol_to_svg (structure) FROM psychoactives;Example output:
<?xml version='1.0' encoding='iso-8859-1'?>
<svg version='1.1' baseProfile='full'
xmlns='http://www.w3.org/2000/svg'
xmlns:rdkit='http://www.rdkit.org/xml'
xmlns:xlink='http://www.w3.org/1999/xlink'
xml:space='preserve'
width='250px' height='200px' viewBox='0 0 250 200'>
<!-- END OF HEADER -->
<rect style='opacity:1.0;fill:#FFFFFF;stroke:none' width='250' height='200' x='0' y='0'> </rect>
<path class='bond-0' d='M 238.636,63.7106 L 204.736,82.0906' style='fill:none;fill-rule:evenodd;stroke:#000000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-1' d='M 204.736,82.0906 L 171.868,61.9221' style='fill:none;fill-rule:evenodd;stroke:#000000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-11' d='M 204.736,82.0906 L 204.306,98.1513' style='fill:none;fill-rule:evenodd;stroke:#000000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-11' d='M 204.306,98.1513 L 203.876,114.212' style='fill:none;fill-rule:evenodd;stroke:#0000FF;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-2' d='M 171.868,61.9221 L 137.968,80.3021' style='fill:none;fill-rule:evenodd;stroke:#000000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-3' d='M 137.968,80.3021 L 105.1,60.1336' style='fill:none;fill-rule:evenodd;stroke:#000000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-3' d='M 129.004,83.8504 L 105.997,69.7324' style='fill:none;fill-rule:evenodd;stroke:#000000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-13' d='M 137.968,80.3021 L 136.935,118.851' style='fill:none;fill-rule:evenodd;stroke:#000000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-4' d='M 105.1,60.1336 L 71.2,78.5136' style='fill:none;fill-rule:evenodd;stroke:#000000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-5' d='M 71.2,78.5136 L 70.1674,117.062' style='fill:none;fill-rule:evenodd;stroke:#000000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-5' d='M 78.7548,84.5024 L 78.032,111.486' style='fill:none;fill-rule:evenodd;stroke:#000000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-14' d='M 71.2,78.5136 L 56.03,73.1314' style='fill:none;fill-rule:evenodd;stroke:#000000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-14' d='M 56.03,73.1314 L 40.86,67.7492' style='fill:none;fill-rule:evenodd;stroke:#FF0000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-6' d='M 70.1674,117.062 L 103.035,137.231' style='fill:none;fill-rule:evenodd;stroke:#000000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-8' d='M 70.1674,117.062 L 54.6783,121.64' style='fill:none;fill-rule:evenodd;stroke:#000000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-8' d='M 54.6783,121.64 L 39.1892,126.218' style='fill:none;fill-rule:evenodd;stroke:#FF0000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-7' d='M 103.035,137.231 L 136.935,118.851' style='fill:none;fill-rule:evenodd;stroke:#000000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-7' d='M 104.444,127.694 L 128.174,114.828' style='fill:none;fill-rule:evenodd;stroke:#000000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-9' d='M 28.775,121.565 L 20.0693,108.882' style='fill:none;fill-rule:evenodd;stroke:#FF0000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-9' d='M 20.0693,108.882 L 11.3636,96.1989' style='fill:none;fill-rule:evenodd;stroke:#000000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-10' d='M 11.3636,96.1989 L 20.6416,84.1227' style='fill:none;fill-rule:evenodd;stroke:#000000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-10' d='M 20.6416,84.1227 L 29.9195,72.0465' style='fill:none;fill-rule:evenodd;stroke:#FF0000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-12' d='M 209.274,124.057 L 222.923,132.433' style='fill:none;fill-rule:evenodd;stroke:#0000FF;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<path class='bond-12' d='M 222.923,132.433 L 236.571,140.808' style='fill:none;fill-rule:evenodd;stroke:#000000;stroke-width:2px;stroke-linecap:butt;stroke-linejoin:miter;stroke-opacity:1' />
<text dominant-baseline="central" text-anchor="middle" x='33.1865' y='129.92' style='font-size:12px;font-style:normal;font-weight:normal;fill-opacity:1;stroke:none;font-family:sans-serif;fill:#FF0000' ><tspan>O</tspan></text>
<text dominant-baseline="central" text-anchor="middle" x='34.8573' y='67.5476' style='font-size:12px;font-style:normal;font-weight:normal;fill-opacity:1;stroke:none;font-family:sans-serif;fill:#FF0000' ><tspan>O</tspan></text>
<text dominant-baseline="central" text-anchor="end" x='207.989' y='122.567' style='font-size:12px;font-style:normal;font-weight:normal;fill-opacity:1;stroke:none;font-family:sans-serif;fill:#0000FF' ><tspan>HN</tspan></text>
</svg>
How many atoms?
SELECT mol_numatoms (structure) FROM psychoactives;How many aromatic rings?
SELECT mol_numaromaticrings (structure) FROM psychoactives;Which compounds contain a specific chemical block?
SELECT * from psychoactives where structure@>'CC1=CC2=C(S1)C(=O)NC(C)=N2'@> : substructure search operator. Returns whether or not the mol or qmol on the right is a substructure of the mol on the left.
<@ : substructure search operator. Returns whether or not the mol or qmol on the left is a substructure of the mol on the right.
@= : returns whether or not two molecules are the same.
< : returns whether or not the left mol is less than the right mol
> : returns whether or not the left mol is greater than the right mol
= : returns whether or not the left mol is equal to the right mol
<= : returns whether or not the left mol is less than or equal to the right mol
>= : returns whether or not the left mol is greater than or equal to the right mol
What's the chemical formula?
SELECT mol_formula (structure) FROM psychoactives;What's the InCHl?
SELECT mol_inchi (structure) FROM psychoactives;Similarity
This is where it gets a little more involved. Read this first: https://en.wikipedia.org/wiki/Chemical_similarity
These are the major fingerprint types:
- Morgan
- Feat Morgan
- MACCS keys
- AtomPair
- RDKit
- Torsion
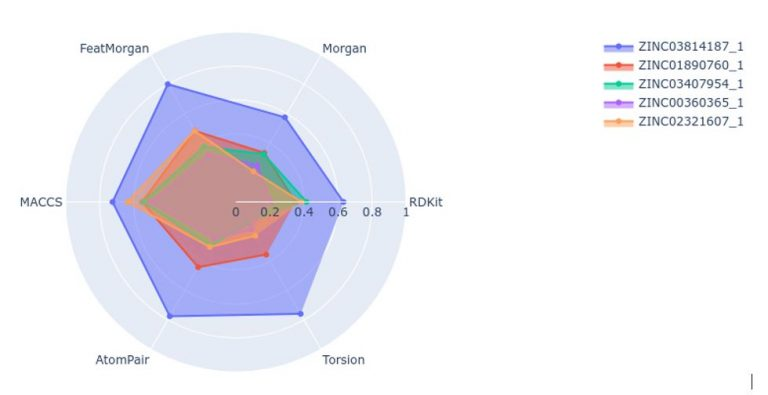
We have two types of similarity in RDKit: Tanimoto similarity and Dice similarity. We're looking for the nearest neighbours of a molecule ebign described in the form of a graph.
They work by creating Morgan Circular Fingerprints (extended-connectivity fingerprint ECFP4, 1965) of molecules from their SMILES string.
The Tanimoto coefficent is determined by looking at the number of chemical features that are common to both molecules (the intersection of the data strings) compared to the number of chemical features that are in either (the union of the data strings). The Dice coefficient also compares these values but using a slightly different weighting.
The Tanimoto coefficient is the ratio of the number of features common to both molecules to the total number of features, i.e.
( A intersect B ) / ( A + B - ( A intersect B ) )
The range is 0 to 1 inclusive.
The Dice coefficient is the number of features in common to both molecules relative to the average size of the total number of features present, i.e.
( A intersect B ) / 0.5 ( A + B )
The weighting factor comes from the 0.5 in the denominator. The range is 0 to 1.
For more on H.L Morgan fingerprinting, see the original 1965 paper "The Generation of a Unique Machine Description for Chemical Structures" at https://pubs.acs.org/doi/abs/10.1021/c160017a018
So to get the 10 compounds most similar to MDMA:
SET rdkit.tanimoto_threshold = 0.6;
SELECT psy_id, structure,
tanimoto_sml (morganbv_fp(mol_from_smiles("CC(CC1=CC2=C(C=C1)OCO2)NC"::cstring)), structure) AS similarity
FROM psychoactives
ORDER BY morganbv_fp(mol_from_smiles("CC(CC1=CC2=C(C=C1)OCO2)NC"::cstring))<%>structure; For non-chemists, this can be translated like so:
- Convert SMILES string "CC(CC1=CC2=C(C=C1)OCO2)NC" to type
mol. - Calculate a bit vector (BV) Morgan fingerprint of the molecule using
morganbv_fp. - Calculate the Tanimoto coefficient of the fingerprint between 0 and 1.
- Order it by the nearest neighbours you find (<%>)
To use Dice, simply change tanimoto_sml to dice_sml.
The result will be 3 fields:
- The primary key (psy_id);
- The SMILES string (structure);
- The similarity score (double precision, from 0 to 1);
% : operator used for similarity searches using Tanimoto similarity. Returns whether or not the Tanimoto similarity between two fingerprints (either two sfp or two bfp values) exceeds rdkit.tanimoto_threshold.
# : operator used for similarity searches using Dice similarity. Returns whether or not the Dice similarity between two fingerprints (either two sfp or two bfp values) exceeds rdkit.dice_threshold.
<%> : used for Tanimoto KNN searches (to return ordered lists of neighbors).
<#> : used for Dice KNN searches (to return ordered lists of neighbors).
How Do We Categorise The Dataset?
Our first step in discovering new molecules is defining what we're looking to predict, recognise, and identify. Ultimately, we're searching for a boolean situation of psychoactive = true | false.
Simple Shulgin Activity
However, we can get better than that. Let's start with the Shulgin Scale, which is based on dosage:
MINUS, n. (-) On the quantitative potency scale (-, ±, +, ++, +++), there were no effects observed.
PLUS/MINUS, n. (±) The level of effectiveness of a drug that indicates a threshold action. If a higher dosage produces a greater response, then the plus/minus (±) was valid. If a higher dosage produces nothing, then this was a false positive.
PLUS ONE, n. (+) The drug is quite certainly active. The chronology can be determined with some accuracy, but the nature of the drug's effects are not yet apparent.
PLUS TWO, n. (++) Both the chronology and the nature of the action of a drug are unmistakably apparent. But you still have some choice as to whether you will accept the adventure, or rather just continue with your ordinary day's plans (if you are an experienced researcher, that is).
PLUS THREE, n. (+++) Not only are the chronology and the nature of a drug's action quite clear, but ignoring its action is no longer an option. The subject is totally engaged in the experience, for better or worse.
PLUS FOUR, n. (++++) A rare and precious transcendental state, which has been called a "peak experience," a "religious experience," "divine transformation," a "state of Samadhi" and many other names in other cultures.
https://en.wikipedia.org/wiki/Shulgin_Rating_Scale
In simple terms, we're looking to discover/predict molecules which will go on to score a Shulgin PLUS ONE (+) threshold or above.
Psychedelic Classification (Subjective Effect Index)
However, we can break this down more. We want to be able to discover the likelihood of specific types of effects. For those, we can turn to PsychonautWiki. The subjective effects of psychoactives can easily be placed into an ontology (a Nested Set: https://en.wikipedia.org/wiki/Nested_set_model).
- Visual Effects
- Auditory Effects
- Tactile Effects
- Disconnective Effects
- Smell & Taste Effects
- Multisensory Effects
- Cognitive Effects
- Physical Effects
- Uncomfortable Physical Effects
See: https://psychonautwiki.org/wiki/Subjective_effects_index
We need to tag our existing data, because we require it to train our AI/ML model(s). Training means to provide a set of records which can be used as a historical reference, that the computer can use to make predictions of what might work in the future. Think of it as a set of correct answers you provide in advance of asking a question.
The answers we want are suggestions for new molecules coupled with a probability/confidence score of their psychoactivity potential ranked ascending from 0.0 to 1.0.
Quantitative Structure–Activity Relationship (QSAR)
Our entire rationale is based on two assumptions:
- The structure of a molecule defines its biological activity, and
- Structurally similar molecules have a similar biological activity.
Each record in our data can be classified with descriptors. Molecular descriptors are mathematical values that describe the structure or shape of molecules, helping predict the activity and properties of molecules in complex experiments.
They are divided into two main categories: experimental measurements, such as log P, molar refractivity, dipole moment, polarisability and, in general, additive physical–chemical properties and theoretical molecular descriptors, which are derived from a symbolic representation of the molecule and can be further classified according to the different types of molecular representation.
Different libraries are available, the best known open source one being Mordred (https://jcheminf.biomedcentral.com/articles/10.1186/s13321-018-0258-y)
For more on QSAR methodology: https://en.wikipedia.org/wiki/Quantitative_structure–activity_relationship
It's worth bearing in mind Dr Zee's wisdom here:
"It’s not nearly as intentional as most people think. The only aspects of a novel molecule I can control are its chemical structure and its legality. But there is no scientific method for predicting what effect it will have on the human body or mind or brain, and whether it will have any effect at all.”
https://www.theguardian.com/tv-and-radio/2016/may/24/dr-zee-the-godfather-of-legal-highs-i-test-everything-on-myself
Setting Up An Offline Home Network For Machine Learning

It's entirely possible to run a home network for machine learning (ML). For this kind of research, you probably don't want it sitting in AWS. Not that research is illegal; more that you'll lose your data.
Processing Power (GPU)
The first thing to know is you're going to need a graphics card (graphics processing unit or GPU) rather than a normal CPU, similar to how you'd need one for mining Bitcoin. The king of GPUs for deep/machine learning is NVIDIA.
According to Tim Dettmers, who recommends the RTX 3080 and RTX 3090:
Definitely buy used GPUs. Used RTX 2070 ($400) and RTX 2060 ($300) are great. If you cannot afford that, the next best option is to try to get a used GTX 1070 ($220) or GTX 1070 Ti ($230). If that is too expensive, a used GTX 980 Ti (6GB $150) or a used GTX 1650 Super ($190).
https://timdettmers.com/2020/09/07/which-gpu-for-deep-learning/
He also says the avoid "any Tesla card; any Quadro card; any Founders Edition card; Titan RTX, Titan V, Titan XP".
You can chain ("parallelise') GPUs into a cluster to improve performance.
If you want something already set up, consider the Lambda Razer TensorBook (https://lambdalabs.com/deep-learning/laptops/tensorbook):

OS Software Setup
Obviously, start with the latest version of Ubuntu and download the latest drivers for your GPU.
Next, to use the GPU, you will need to install CUDA (Compute Unified Device Architecture) for parallel computation.
Optionally, you can install cuDNN (NVIDIA CUDA Deep Neural Network) for advanced calculations.
Next, you need your specialist ML software:
- Keras deep learning framework (https://keras.io/)
- PyTorch machine learning framework (https://pytorch.org/)
- TensorFlow ML model framework (https://www.tensorflow.org/)
- JupyterLab data notebook (https://jupyter.org/install)
Optionally, pick up some other open-source software:
- Apache Mahout (https://mahout.apache.org/)
- Apache MXNet (https://mxnet.incubator.apache.org/versions/1.9.1/)
- Apache Zeppelin (https://zeppelin.apache.org/)
- Mathworks MATLAB (https://www.mathworks.com/solutions/deep-learning.html)
- Chainer (https://chainer.org/)
- RStudio IDE (https://www.rstudio.com/)
- Microsoft Cognitive Toolkit CNTK (https://learn.microsoft.com/en-us/cognitive-toolkit/)
- RapidMiner (https://rapidminer.com/)
Understanding Machine Learning (ML)
Drug discovery using cheminformatics isn't new. It's a HUGE field with enormous potential, which is being chased by multi-nationals and medtech companies all over the world. The literature is extensive.
Generally in AI/ML, we are looking to automate a number of different things:
- Classify something (apply a label, categorise etc)
- Rank a list of things (score, order etc)
- Make a decision about something
Explaining all the intricacies of deep machine learning is far beyond the scope of this article, but if you're new, the following is an overview of the main components.
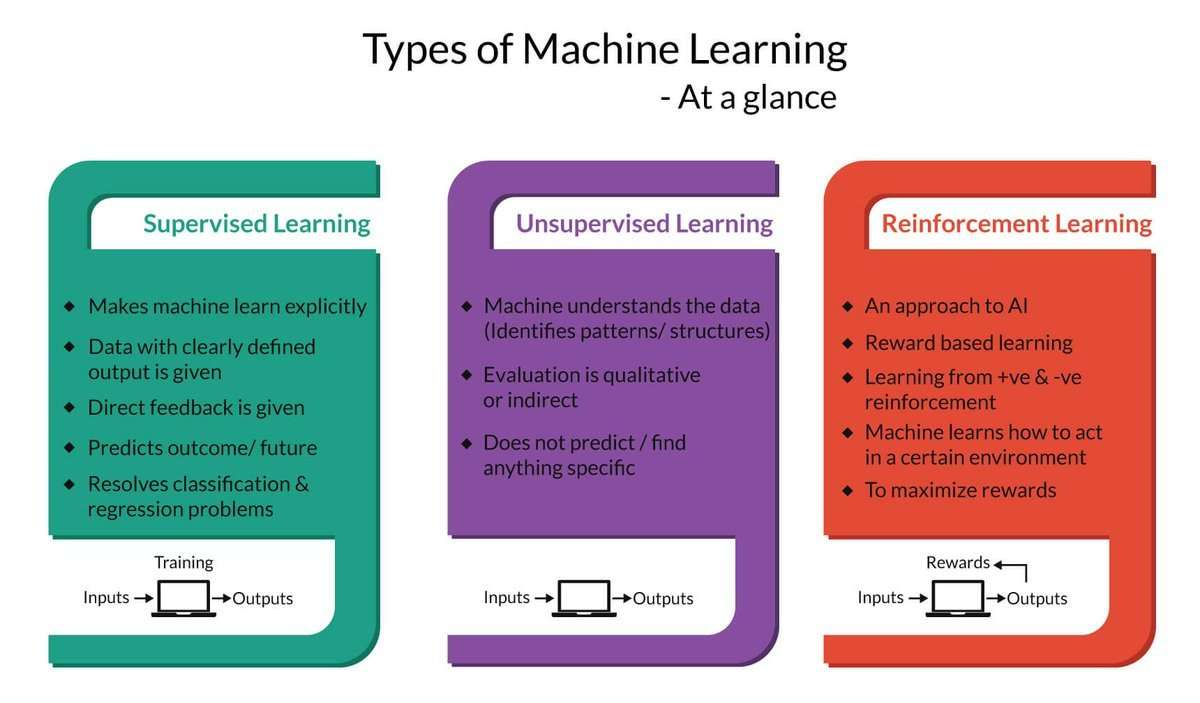
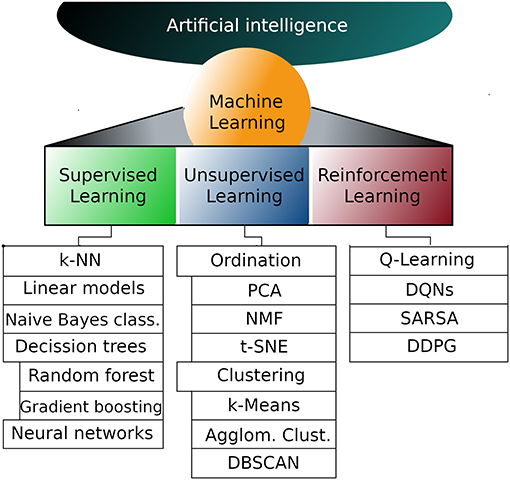
But first, let's take a look at the existing work. The process of machine learning broadly falls into 3 categories.
Supervised Learning
A labeled dataset acts as a trainer, teaching the model or the machine. Once trained, the model can begin making predictions and decisions as new data is received. It can also be further grouped into regression (when the output variable is a real value, such as “dollars” or “weight”) and classification (when the output variable is a category, such as “red” or “blue” or “disease” and “no disease”) problems.
Example: is the image of a car or a plane? A cat or a dog?
- Nearest Neighbor
- Naive Bayes
- Decision Trees
- Linear Regression
- Support Vector Machines (SVM)
- Neural Networks
This is where most chemistry models to date tend to focus.
Unsupervised Learning
This identifies the relationships or patterns in unlabeled data. The model learns independently (without a trainer) through observation and creates clusters of the observed patterns and relationships in the dataset. There are no correct answers and there is no teacher. It can be further divided into clustering (inherent groupings in the data, such as purchasing behavior) and association (people that buy X also tend to buy Y) problems.
Example: What is the likelihood this financial transaction could be grouped with other fraudulent transactions?
- Apriori
- Equivalence Class Clustering and bottom-up Lattice Traversal
- frequent pattern (FP) growth
- K-means clustering
- Principal component analysis (PCA)
Sequential (Reinforcement) Learning
This allows an agent, which is a goal-oriented entity, to learn in an interactive environment using feedback from its own actions and experiences. Sequential learning relies on trial and error to make a sequence of decisions.
Example: A piece of text in English translated into French.
- Q-Learning
- Temporal Difference (TD)
- Deep Adversarial Networks
A great start to reading up on the background of this tech is "Deep Learning For the Life Sciences".

The process is much the same:
- Get your training dataset as a CSV file (my_data.csv);
- Load CSV data into your Jupyter notebook;
- Write Python code to produce a set of molecular descriptors (traits or characteristics of each record, e.g. molecular weight, number of atoms etc);
- Write Python code to build a training model, which is applying an algorithm to the data;
- Split the data into two subsets: training, and testing;
- etc etc
Drug Discovery AI/ML Theory (So Far)
So how do we approach this for the process of discovering new substances? It's a febrile niche which is changing rapidly.
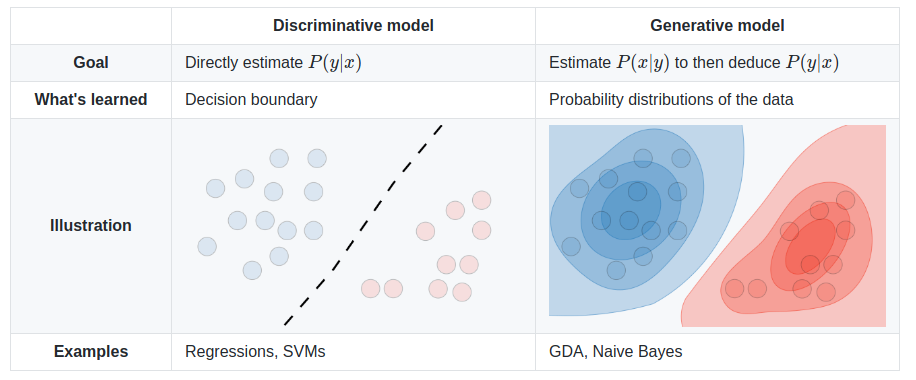
We can do two types of things: discriminate between records, or generate new ones. A generative model could generate new photos of animals that look like real animals, while a discriminative model could tell a dog from a cat. A generative model is a type of AI architecture that generates new data that is similar characteristically to the training data.
The literature so far focuses on data we already have, i.e. molecules we have already theorised on the whiteboard.
For a good high-end overview:
- https://www.sciencedirect.com/science/article/pii/S2001037021003421
- https://machinelearningmastery.com/a-tour-of-machine-learning-algorithms/
First, let's look at some example notebooks:
Open Drug Discovery Toolkit (ODDT) is modular and comprehensive toolkit for use in cheminformatics, molecular modeling etc. ODDT is written in Python, and make extensive use of Numpy/Scipy
See: https://github.com/oddt/oddt
Also see: https://github.com/DeepGraphLearning/torchdrug
DeepChem aims to provide a high quality open-source toolchain that democratizes the use of deep-learning in drug discovery, materials science, quantum chemistry, and biology.
https://github.com/deepchem/deepchem
This repository contains notebooks that will guide you through the process of data preparation and model training to predict chemical properties of compounds represented as SMILES.
See: https://github.com/Shiska07/Cheminformatics-and-Drug-Discovery
End-to-end active learning pipeline for virtual screening and drug discovery. The datasets used in this study are: PriA-SSB target, 107 PubChem BioAssay targets, and PstP target.
See: https://github.com/gitter-lab/active-learning-drug-discovery
Certain algorithms have been hereto more popular than others, and most are discriminative.
Naive Bayes
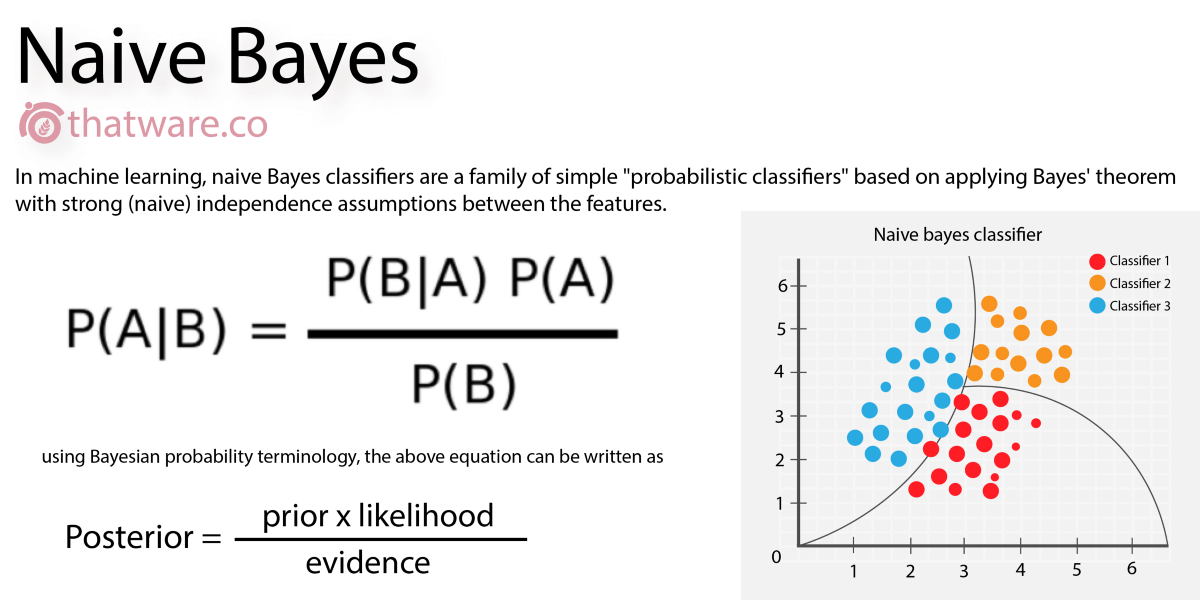
A classification algorithm which works on the basis of probability, given a set of features. For example, given 7 days with different weather (as a "feature" of the day), we can classify the day as good for playing golf, or not. In drug discovery, we can describe the "features" of a molecule, and determine a yes/no to a question. It's known as "naive" because one thing is (naively) assumed not to affect any other thing.
- https://www.geeksforgeeks.org/naive-bayes-classifiers/
- https://www.tutorialspoint.com/machine_learning_with_python/classification_algorithms_naive_bayes.htm
Support Vector Machine
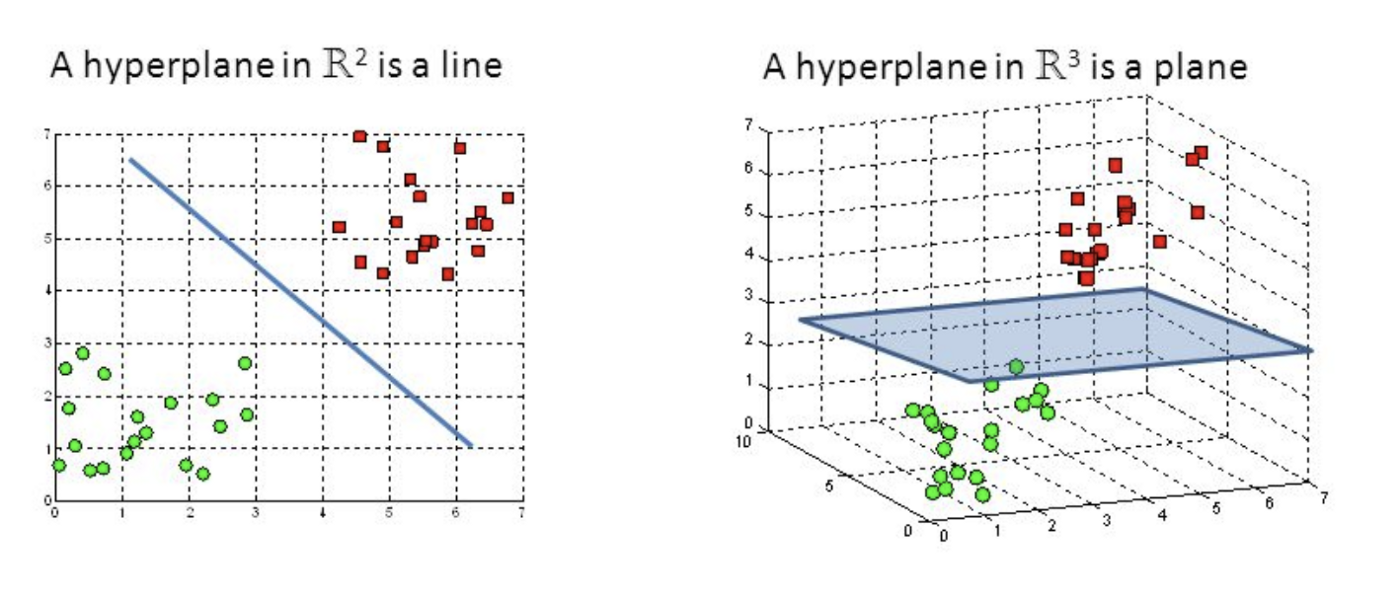
Also a classification algorithm which splits a dataset into two groups (a binary yes/no), via a line on a graph (aka a "hyperplane") which functions as a decision boundary. Group A may be the most likely candidates. We can combine many of them to perform Multiple Kernel Learning (MKL).
- https://www.geeksforgeeks.org/introduction-to-support-vector-machines-svm/
- https://www.tutorialspoint.com/machine_learning_with_python/classification_algorithms_support_vector_machine.htm
Random Forest
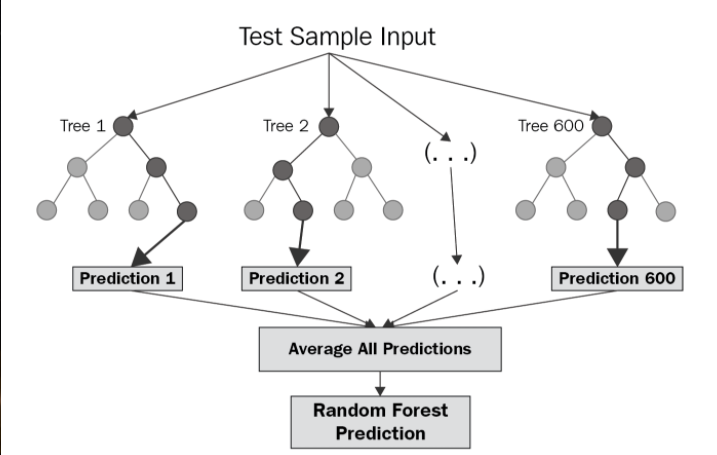
A joint classification/regression algorithm which works by bagging and/or boosting the output of multiple decision trees as a "forest". By combining information about molecules and answering many questions at once, we create a master decision tree.
- https://www.geeksforgeeks.org/random-forest-regression-in-python/
- https://www.tutorialspoint.com/machine_learning_with_python/classification_algorithms_random_forest.htm
Generating Novel Molecules
How far can we go with this approach? A nice beginning a visit to the ChEMBL database (https://www.ebi.ac.uk/chembl/) and this project published from the Royal Society of Chemistry:
Several recent ML algorithms for de novo molecule generation have been utilized to create an open-access database of virtual molecules. The algorithms were trained on samples from ZINC, a free database of commercially available compounds. Generated molecules, stemming from 10 different ML frameworks, along with their calculated properties were merged into a database and coupled to a web interface, which allows users to browse the data in a user friendly and convenient manner.
"cheML.io: an online database of ML-generated molecules"
https://pubs.rsc.org/en/content/articlehtml/2020/ra/d0ra07820d
Demo: https://cheml.io/
What we want as our output is a list of SMILES strings, which represent completely new molecules with psychoactive properties similar to things we have seen before. For this, we need to use a generative approach.
Rather than theorise them manually, we want our model to guess a list of potential candidate molecules which aren't found in any existing DB, ranked by the probability they are psychoactive.
Example: given a SMILES string, such as CC1=CC=C(C(C(NC)C)=O)C=C1 (Mephedrone, or 4-methylmethcathinone), the generative output resembles:
+-----------------------------------+-------+
| Candidate | Score |
+-----------------------------------+-------+
| CC(C(=O)C1=CC=C(C=C1)OC)NC | 0.91 |
| CCNC(C)C(=O)C1=CC=C(C=C1)C | 0.85 |
| CC1=CC=C(C=C1)C(=O)C(C)N2CCCC2.Cl | 0.76 |
+-----------------------------------+-------+(These would be the actual results of a similarity search, but are here to illustrate the target input/output operation).
IBM likes this so approach much it released their Generative Toolkit for Scientific Discovery ("an open-source library to accelerate hypothesis generation in the scientific discovery process"): https://github.com/GT4SD/gt4sd-core
There are 7 main algorithms:
- Autoregressive Models
- Bayesian Network
- Generative Adversarial Networks (GAN)
- Gaussian Mixture Model
- Hidden Markov Model
- Latent Dirichlet Allocation (LDA)
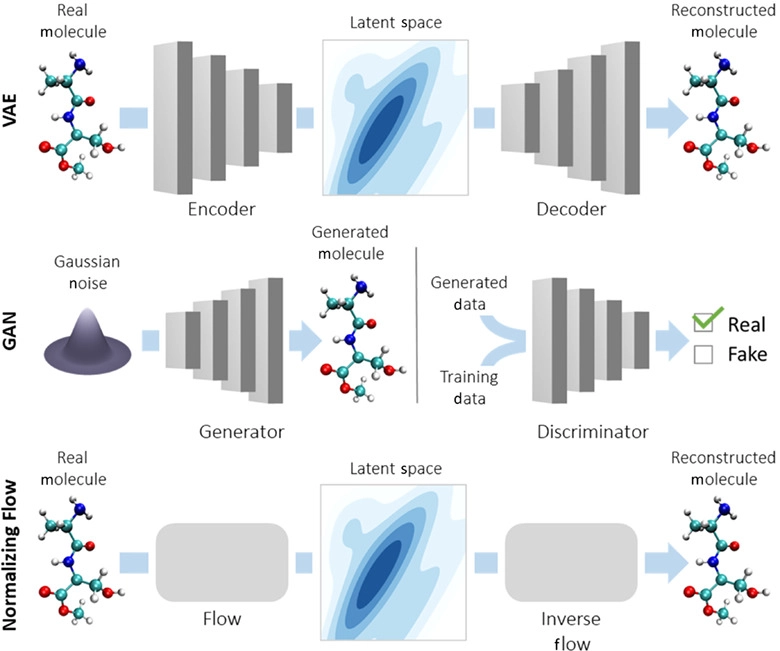
- Variational Autoencoders (VAEs)
More:
- https://www.sciencedirect.com/science/article/pii/S1359644621002531
- https://www.researchgate.net/publication/354370677_Generative_AI_Models_for_Drug_Discovery
- https://wires.onlinelibrary.wiley.com/doi/full/10.1002/wcms.1608
The last one - VAEs - has been used extensively to generate artwork and human faces.
Variational Autoencoders (VAEs) have been one of the most popular approaches to unsupervised learning of complicated distributions. They are built on top of standard function approximators, which are neural networks and can be trained with stochastic gradient descent. The application of VAEs includes generating various kinds of complicated data, including handwritten digits, faces, CIFAR images, predicting the future from static images and more.
Joey Mach explains a fascinating way to use it for SMILES target discovery:
In the scenario of generating molecules, the input for the GRU or the decoder is a vector representing the compressed form or latent space representation of the molecules. The final output of the GRU is the reconstruction of the molecule in its original/expanded representation. The quantity which measures the drug-likeness of a molecule is called QED (quantitative estimation of drug-likeness).
All generated molecules with a QED below 0.5 are disregarded, this ensures the quality of the generated molecules.
- https://towardsdatascience.com/unlocking-drug-discovery-through-machine-learning-part-1-8b2a64333e07
- https://github.com/joeymach/Leveraging-VAE-to-generate-molecules
There are plenty more ideas on the way.
The AI-Powered Underground Drug Lab Of The Future
Hopefully, most of this dry theory has imprinted on your brain the entire futility of criminalising certain areas of chemistry. When it's open source, it's for everyone. Clever legislisation like the US Federal Analogue Act, 21 U.S.C. § 813 and the UK Psychoactive Substances Act 2016 is never going to stop the discovery of new substances, even if it may suppress their production and distribution.
Now AI is for everyone, because it's open source, it means drug discovery is for everyone. That means psychoactives and "illegal" chemistry. A criminal gang could produce the computational power needed easily, and/or employ the technicians for twice their Silicon Valley salary. Particularly as the intellectual research itself is protected speech and a perfectly legal activity.
Moreover, what can be done by law enforcement if a raid on a lab only yields a bunch of computer equipment which has already published its results onto a decentralised network like ZeroNet?
What happens when a gang invests $5M into a Colombian data center which produces 2500 new AI-generated psychedelics? And another rival does the same?
The cat-and-mouse game is over. There is no possible way any law enforcement agency or global watchdog would be able to keep up.
The Patient-Zero for this is "M", who created the Ketamine analog, Methoxetamine.
After receiving a bachelor’s degree in biochemistry I was working on my masters in neuropharmacology. I synthesized an array of phenmetrazine analogs and quantified their potency as anorectics.
I taught neurobiology as part of my postgraduate degree. But then I transitioned from academia into the sort of independent research I’m doing now.
The methoxetamine molecule was something I had floating around in my head for about three years. I just knew it would really be something fantastic; it contains every necessary functional group to produce the perfect dissociative. I felt it would be like a stress-free version of ketamine. Eventually I found someone who was interested and made a small batch.
https://www.vice.com/en/article/ppzgk9/interview-with-ketamine-chemist-704-v18n2
The process is artistic and creative. You can no more suppress a scientist's need to experiment than you can a musician's need to write a song. There's no way to win this battle.
An astute network of chemists would recognise:
- The need for a comprehensive, downloadable open source database of discovered psychoactives, with each record tagged with its molecular classifiers (a "PubChem for psychoactives");
- The need to use generative ML models for creating drug candidates, and their synthesis routes;
- The need to use discriminative ML models to filter/flag candidates on their basis for biological activity, and routes for complexity and precursor availability;
- The need to share data to reduce harm.
Law enforcement had enough trouble keeping up with Shulgin or Nichols' labs. The endgame for synthetics is very much in sight, and the result is foregone: the authorities lose. No-one should celebrate that, because these (mainly good) people are the same as those picking up the overdose victims to put them in the ambulance.
Imagine fifteen labs around the world synthesising ten new drugs a month from 300 AI-generated SMILES strings. That's 4500 "potentials" published every month and 1800 new items for sale a year.
It's the same as a magnet:// link for Bittorrent. A text link, smiles://CC(NC)CC1=CC=C(OCO2)C2=C1 which can be copied/pasted isn't something anyone can control.
Now imagine if they shared that information. 54,000 records a year, or a few megabytes in a text file.
The answer is politically painful: regulate a market. But we won't get to that until we are so swamped in analogs, governments admit they cannot cope. Which hasn't exactly been a forthcoming sentiment in the War on Drugs so far.